Selection
Simple genomes
Complex genomes
Cloning DNA
Sequencing
Selection
In the wild, each species may exist as one population or multiple populations. Different populations correspond to defined areas – habitats.
The sum of all present alleles for a given gene in a given population is known as the gene pool.
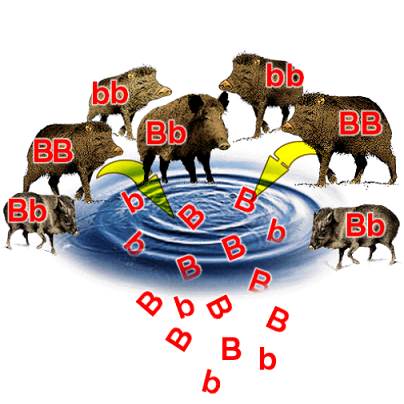
This is essentially a way of thinking about all the individuals in a population contributing their alleles towards the overall allele frequency. The extent of different alleles present gives the genetic diversity of a population.

The allele frequency in a population’s gene pool can change as a result of selection. The effectors of selection can be varied, yet the outcome is similar: advantageous or preferred alleles and the traits associated with them increase in frequency, while detrimental or disfavoured alleles and the traits associated with them decrease in frequency.
Here is an all-time classic example. The most frequent initial moth colour in a population landing on tree trunks was dark, to match that of the tree trunks. Few moths could get away with being light-coloured. Once the tree trunks were painted white, the former moths became very apparent to predators, and so the light-coloured moths evaded predation much better and survived to reproduce. Essentially, the tables had turned!
This resulted in the allele for light colour to spread and become the most frequent compared to that for dark colour. The latter sharply dropped in frequency and became the minority.


This is an example of directional selection. It tends towards an extreme, either the light-coloured or the dark-coloured, depending on scenario.
Selection can also tend towards a “happy medium” and avoid either extreme. This is stabilising selection. If really small..