Health and Safety
Liquids and Solutions
Separation Techniques
Electrophoresis
Antibody Techniques
Microscopy
Aseptic Technique and Cell Culture
Health and safety
Much biological research relies on the use of chemicals and organisms. Some of these are an inherent health and safety hazard. For example, most pure chemicals are irritants in certain doses, while others such as organic solvents and fungicides can be straight-up lethal to humans even in small doses.
This is why labs are run with a clear health and safety framework in place. This varies from lab to lab, depending on the research being carried out, and can include security clearance access to certain parts of the lab, designated first aid officers, clothing and personal protective equipment (PPE) guidelines and supervision systems.


For example, lab coats and nitrile gloves are standard procedure in most labs, and in more chemistry-focused labs, protective goggles too.
Beyond the risks present to lab workers, some items used in labs can be dangerous to other organisms and the environment once leaked into the outside world. Some chemicals will kill aquatic life or other animals, even if safe for humans. Therefore, depending on the usage of these chemicals, some labs will have a building-wide implementation of waste disposal and filtering as well as chemical dilution practices to minimise this risk.
Commonly, waste is incinerated to remove the risk of these materials reaching public spaces and the natural environment, whether they are toxic chemicals or genetically modified bacteria.
Lipids and solutions
Lipids and solutions are prepared in the lab as nutrients for growing microorganisms, buffers to hold proteins in their correct folding, chemical solutions to undergo various reactions, and anything else one’s mind can think up. In the lab setting, even plain water needs to be pure, and pH is measured to one decimal point. Liquids can be handled down to each individual tenth of a microlitre, which is a 10,000,000th of a litre.
A common task when handling liquids in an experiment is the need to dilute a stock into multiple serial dilutions with decreasing concentration of the solute in the solvent i.e. glycerol in water. For example, 5 tests are undertaken, each a tenth of the concentration of the original solution. So a 50% glycerol solution of 10 mL (which contains 5 mL of glycerol and 5 mL of pure water) is used to create the subsequent 4 solutions.
In this serial dilution, a tenth (i.e. 1 mL) of the original solution is added to a new tube, and made up to 10 mL with 9 mL of pure water. This is now a 5% glycerol solution. A tenth of this is used to make the next solution of 0.5% glycerol, and so forth. This is a log dilution series because each dilution is a factor (in this case of 10) less concentrated that the previous.
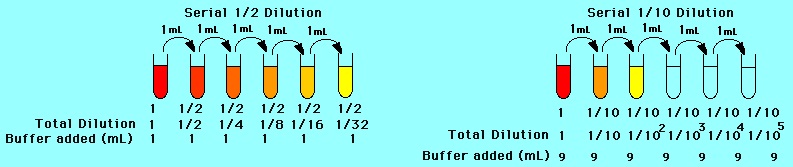
Depending on how dilute we need the solution……