Cloning DNA with PCR
Visualising DNA with gel electrophoresis
Genetic fingerprinting technique
Using DNA
Genetically modified organisms (GMOs)
Super soya beans
Cloning DNA with PCR
If we have obtained a DNA sample or a few, what next? Well, nothing much can be done with that. We must obtain exponentially more DNA to use for any purpose. And it all of course must be identical. We must essentially clone our DNA. Considered the very staple of molecular biology, this technique for multiplying DNA many-fold was invented by a chap Kary Mullis who believes in astrology.
Essentially the DNA is denatured so the 2 strands break apart, short complementary bits called primers attach to the strands, the enzyme DNA polymerase binds to the primers and initiates the assembly of a new DNA strand, and finally the process is repeated many times over in a chain reaction. This is the polymerase chain reaction, PCR.
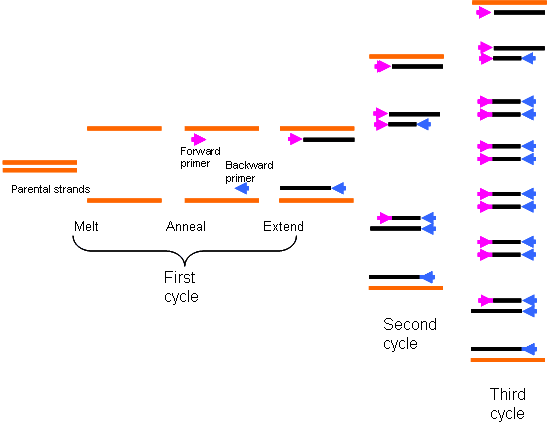
Soon enough, the few bits of DNA become thousands, and hundreds of thousands, and millions…
Visualising DNA with gel electrophoresis
A common method of visualising differences is gel electrophoresis which involves loading small volumes of samples on a gel and running a current across it in order to separate the samples by…