Introduction
Basics
Chi-squared test
Mutation
The cause of mutations
The effect of mutations
Sickle cell anaemia
Cystic fibrosis
Phenylketonuria (PKU)
Huntington’s disease
Sex linkage and the autosomal linkage
Investigating inheritance with model organisms
Down’s syndrome, Turner’s syndrome and Klinefelter’s syndrome
Chromosomal mutations
Introduction
If you crossed a green pea plant with a yellow pea plant, what colour would the offspring be? What about their offspring’s offspring?
This is precisely what you’ll be able to answer by the end of this topic.
Basics
Versions of the same gene that give rise to different phenotypes are called alleles. For example, the gene responsible for ear shape in a cat may have 2 alleles: pointy shape and oval shape.
One of these alleles may be expressed at the expense of another, where both are present together in a cat. Say that the oval shape allele is dominant while the pointy shape is recessive.
Because cats also have 2 copies of each chromosome (and therefore gene), these alleles are written as 2 letters.
If the gene for ear shape is abbreviated as E or e for ear, then the alleles would be:
E (dominant) for oval shape and
e (recessive) for pointy shape
These upper case – dominant, lower case – recessive notation rules are used universally. So what combinations may be present in a cat?
EE, ee or Ee
So what happens in crossing?
EE x ee gives rise to 4 combinations: Ee, Ee, Ee and Ee! 100% heterozygous where the cats will appear oval-ear shaped, yet also carry the recessive allele for pointy ears. Let’s do a second cross.
Ee x Ee gives rise to 4 combinations: EE, Ee, eE and ee. That’s 50% homozygous and 50% heterozygous. 3/4 will have oval ears while 1/4 will have pointy ears.
The first two examples are called homozygous because the same allele (E or e) is present twice, while the last example is heterozygous because different alleles (E and e) are present.
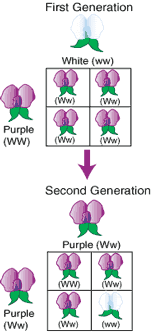
Now see what happened in Mendel’s pea plant experiments?
It’s also possible to have multiple….