Practical skills assessed in the practical endorsement
Independent Thinking
Use and Application of the Scientific Method and Practices
Obtaining Results
Evaluation of Results
Research and Referencing
Instruments and Equipment
Chromatography
Electrophoresis
Using Plants and Animals safely and Ethically
Microbiology aspect Techniques
Fieldwork Sampling Techniques
Data Processing
Independent thinking
This refers to you having the knowledge and confidence to approach a problem or experiment with common sense. It refers to you taking it upon yourself to understand the background, what is going on and what you might be able to do. Ask questions, find out information, update it as you go along. It’s not a test to trip you up, simply material with which you can toy.
Looking things up that you don’t understand is a fundamental unit of not just the A level science, but research itself at the highest level. Research literally means looking things up. Having no clue what is going on is the default. You are not learning random bits of stuff, you are learning how to learn anything.
And how to learn anything requires independent thinking. Binge on it!
Applying investigative approaches refers to the general pattern of findings things out and following up on them. It is part of the overarching cycle of the scientific process.
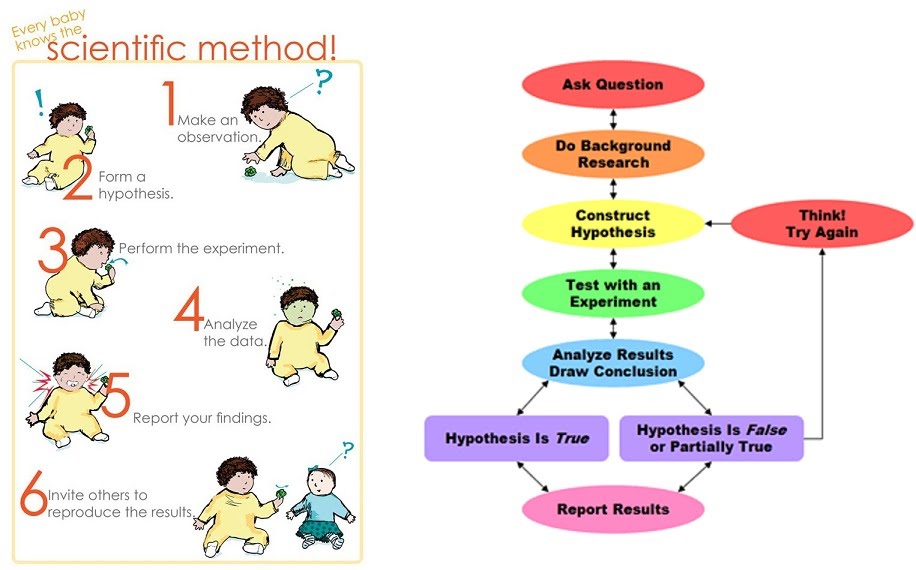
Use and application of scientific methods and practices
The method
Science provides models for harnessing knowledge in a relatively objective, material and quantified environment. It involves high levels of reproducibility and peer-to-peer confirmation of findings, and as such can produce very widely applicable and powerful knowledge. The downside to this is that due to its basis of formulating testable hypotheses, many types of knowledge remain outside of the working field of science.
Starting out with an idea based on previous knowledge or new observation, a testable hypothesis is established as the foundation for experimentation. Hypotheses are statements to be tested. For example, “cats don’t have a food preference” is a testable hypothesis for an animal shelter in the UK, but would not be a testable hypothesis somewhere where there are no cats. A hypothesis might not be testable due to abstract constraints e.g. “people are not happier on the Moon than on Earth”, or due to the limitations of human existence at a given time e.g. time, money, politics, priorities, taboos, etc.
The default hypothesis in any case is the null hypothesis that states no change will be observed. For example, “cats don’t have a food preference for wet or dry food” is the null hypothesis. “Cats prefer wet food to dry food” is its counterpart alternative hypothesis. A statistical test on data obtained from experiments might show that the null hypothesis is to be accepted or rejected.
Once within the space of a testable hypothesis, experimental design follows. This is a preparatory exercise ahead of experimentation that ensures the experiments and outcomes are what they need to be. Experiments must adhere to guidelines such as risk assessment, reproducibility and validity or results, time and cost effectiveness, etc. For example, in a clinical trial where clinicians administer drugs and placebos randomly to patients, a double-blind experimental design is required where neither the clinicians nor the patients are aware of whether they are assigned the drug or the placebo.
Experimental design covers the equipment and reagents needed and whether these are safe and cost-effective enough to justify their use; what experiments will be carried out, when, in what order, how and how many times; what data will need to be recorded; what biases might arise and how to counteract them e.g. labelling tests using codes rather than content names; how to organise experiments to fit with experimenter’s schedules or equipment booking schedules; how to collect the right amount of data from experiments to be able to use certain statistical tests afterwards i.e. some tests need a minimum of data to apply.