Natural selection
Genetic drift
The founder effect
Genetic bottlenecks
The Hardy-Weinberg principle
Natural selection
In the wild, each species may exist as one population or multiple populations. Different populations correspond to defined areas – habitats.
The sum of all present alleles for a given gene in a given population is known as the gene pool.
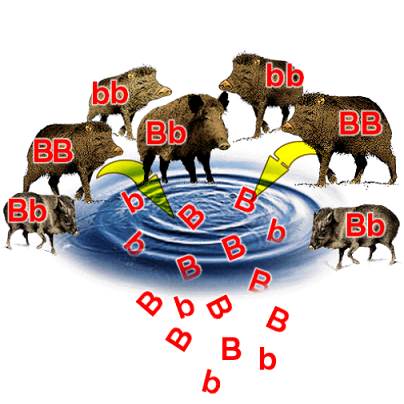
This is essentially a way of thinking about all the individuals in a population contributing their alleles towards the overall allele frequency. The extent of different alleles present gives the genetic diversity of a population.

The allele frequency in a population’s gene pool can change as a result of selection. The effectors of selection can be varied, yet the outcome is similar: advantageous or preferred alleles and the traits associated with them increase in frequency, while detrimental or disfavoured alleles and the traits associated with them decrease in frequency.
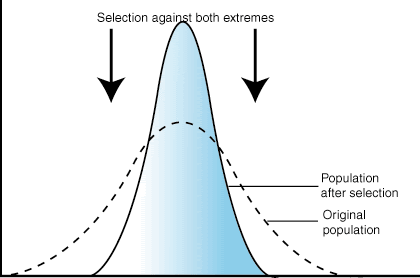
Selection can tend towards a “happy medium” and avoid either extreme. This is stabilising selection. If really small lions don’t survive long, but really large lions can’t supply themselves enough food, then the average lions are selected for and achieve the highest frequency.
There is another type called disruptive selection. Instead of shifting the traits towards a middle ground, disruptive selection splits the….