Polymerase chain reaction
Controls and applications
Polymerase chain reaction
Soon enough, the few bits of DNA become thousands, and hundreds of thousands, and millions…
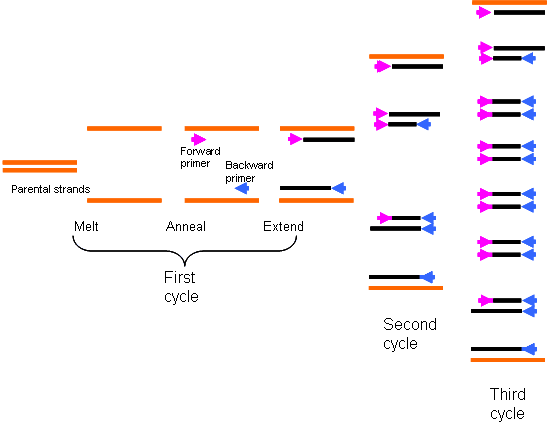
The components of PCR can fit in a very small tube which is placed in a specialised thermocycler or water bath in order to expose it to these fluctuating temperatures. Thermocyclers can be programmed to run automatically on a cycle along the lines of (degrees Celsius) 90-60-70 each for a few minutes, repeated many times over e.g. 30 times. Overall, this can take a few hours to complete.
The fluctuations in temperature correspond to each step in PCR. The highest temperature is required to separate the strands. The lower, annealing temperature bring the strands closer again, in order to bind the primers required to kick-start replication by DNA polymerase, while the temperature lower than the denaturing step, but higher than the annealing step is required for the addition of nucleotides by the polymerase – extending.
These temperatures are well above most physiological conditions where enzymes like polymerase would be functional, so special polymerases are used in PCR which are heat-resistant. They were isolated from microorganisms found living in hot springs and such extreme environments.
Other ingredients of PCR include the nucleotides themselves (free and ready to be added to new DNA strands by polymerase), other optimising agents such as magnesium ions for the DNA polymerase, and water.