Proteome and protein structure
Amino Acids
Primary, Secondary, Tertiary and Quaternary Structure
Collagen and Haemoglobin
Temperature
pH
Protein placement
Plasma membrane
Interaction and Regulation
Inhibitors
The proteome and protein structure
The complete set of genes in a cell is known as the genome while the full range of proteins a cell can make is the proteome. The proteome can be many times larger than its corresponding genome due to multiple arrangements of the mRNA product (alternative splicing) as well as post-translational modifications that can tweak the final protein in different ways based off the same polypeptide.

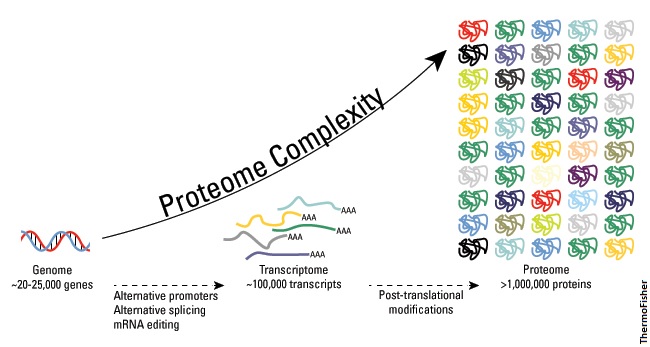
So for humans, a rough 20,000 genes are enough to spawn more than a million different proteins.
Proteins are at the heart of living organisms. Their functions are very varied, from the hair on your head, to the haemoglobin in your red blood cells (which carries oxygen around the body), to the claws of a lion, to insulin (blood glucose regulation). All these highly varied polymer proteins are made of their building blocks, monomers called amino acids.
This is what the generalised structure of an amino acid looks like (make sure you can draw this):
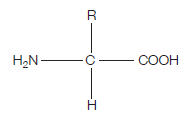
If you’re wondering what this actually is, read on. The clues are in the name (as they usually are).
AMINO – the H2N on the left hand side is an amino group.
ACID – the COOH on the right hand side is a carboxylic acid group (simply an acid)
The hydrogen (H) on the bottom is there all the time (just like the amino group and the acid group), while the R group is the variable which determines what particular amino acid this will be. For example, if the R group was a hydrogen, the amino acid would be glycine.
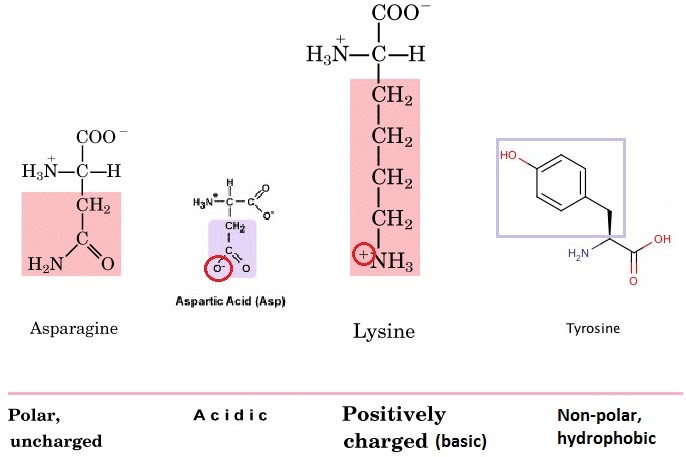
The R group (functional group) on the amino acid also determines the characteristics of that amino acid, including whether it is basic or acidic, polar or hydrophobic. Amino acids can be charged (positively or negatively) or uncharged, polar or non-polar. On asparagine, for example, it is the extra carboxyl (CO) group that makes it polar….