Aseptic technique
Yummy food
Measuring growth
Growing microorganisms has been a fundamental element of much of experimental biology, as well as the underpinning of many modern molecular biology techniques. Perhaps we have a sample of earth that we want to analyse to find a new microorganism with antibacterial properties. Perhaps we are testing a patient sample for an infectious agent. Most likely, we are culturing a safe strain of E. coli that has been genetically modified to produce a protein of interest like human insulin that we can isolate from it and administer to patients.
Aseptic technique
Aseptic means free of contamination. There are hundreds of fungal spores in the air we breathe at all times. There are bacteria and viruses everywhere. If we are to culture Escherichia coli (bacteria, prokaryote) or perhaps Pichia pastoris (yeast, eukaryote), we’re going to be feeding them some nice nutrients, and chances are, loads of other microorganisms will jump at the opportunity to feast.
We don’t want contamination, we just want our specific species that we are culturing and nothing else. The various techniques employed to this end have evolved through time and can even differ between labs and scientists:
A flame (Bunsen burner) can be used in the close vicinity of handling the target microorganism and related equipment and reagents, in order to make the surrounding air warm up and rise higher, carrying away any contaminants that might be present in the air close to our working space.
The equipment we use can be sterile as bought (e.g. plastic loops in sealed bags) or sterilised by passing it through the flame after dipping it in ethanol (e.g. reusable metal loop). Similarly, the lids and necks of bottles of liquids can be passed through the flame briefly upon opening and closing.
The working area can be cleaned with a 70% ethanol (now slightly changed and called IMS-industrial methylated spirit to make it unfit for human consumption) solution before and after the procedure is done, and to clean any other items as necessary, such as gloves, other items and surfaces, etc.
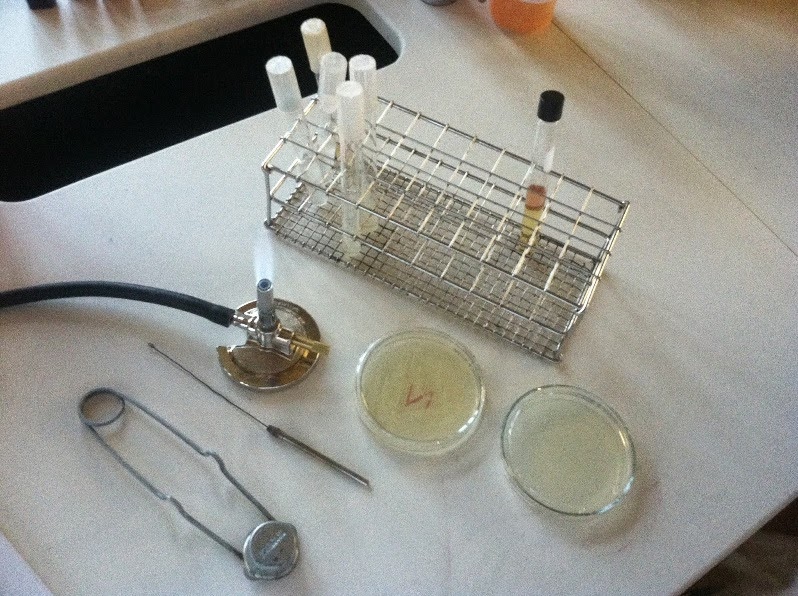
Sidenote: what even is that giant safety pin???
A step up from using a flame is using biological safety cabinets that provide a larger, fully controlled and enclosed working area, which filters the air mechanically to maximise safety and minimise contamination. This also needs to be maintained sterile with ethanol and other cleaning agents, and all samples and equipment kept inside must be separately sterilised with the ethanol solution as they are being used, taken in and out…